Genetic inheritance
The genetic material that codes for various traits lie on DNA wrapped up in chromosomes, in the cell nucleus. Sometimes traits are controlled by a single gene (e.g red and black Gouldian finch head colour), or multiple genes with small, cumulative effects (e.g. body size, clutch size). All mammals and birds are "diploid" (2n) meaning that every individual has a copy of a chromosome inherited from their mother, and one from their father.
To create sperm and eggs (that form a diploid zygote), germ-line cells go through meiosis (Figure 1). Meiosis ultimately creates 4 daughter cells (sperm or eggs) that are haploid (n) - containing a single chromosome. During meiosis, homologous chromosomes pair up and exchange genetic material in a process known as crossing over (Figure 1). Homologous chromosomes are those that inherited from each parent and contain the same sets of genes (e.g both contain the gene for blue eyes). Crossing over shuffles portions of the chromosomes so that the sperm and eggs have novel combinations of genes.
To create sperm and eggs (that form a diploid zygote), germ-line cells go through meiosis (Figure 1). Meiosis ultimately creates 4 daughter cells (sperm or eggs) that are haploid (n) - containing a single chromosome. During meiosis, homologous chromosomes pair up and exchange genetic material in a process known as crossing over (Figure 1). Homologous chromosomes are those that inherited from each parent and contain the same sets of genes (e.g both contain the gene for blue eyes). Crossing over shuffles portions of the chromosomes so that the sperm and eggs have novel combinations of genes.

Figure 1: Meiosis is the formation of haploid gametes (egg and sperm) from diploid germ-cells. Pairs of homologous chromosomes are paired and swap genetic material - to create new genteic combinations on the chromosomes.
Source: https://hscbiology.wordpress.com/blueprint-of-life/
Relatedness & relatedness coefficients
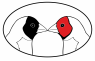
 Figure 2: The genetic material shared by half-siblings
Source: http://genetics.thetech.org/ask-a-geneticist/inherited-parental-dna-two-half-siblings
Figure 2: The genetic material shared by half-siblings
Source: http://genetics.thetech.org/ask-a-geneticist/inherited-parental-dna-two-half-siblings
Because offspring inherit one chromosome from their mother and one from their father, a parent and offspring share 50% of their genetic material. Similarly, siblings that share the same parents will share on average 50% of their genetic material. However, due to the process of crossing over in meiosis, siblings will can share more (or less) than 50% of their genetic material.This logic can be extended to other relatives, such as half-siblings. Half-siblings share only a single parent, and share (on average) 25% of their genetic material. Identical twins or clones share 100% of their genetic material.
The coefficient of relatedness (r) is expressed as the probability that two individuals will share the same allele due to recent common ancestry (e.g. they shared parents). It can also be thought of as the proportion of the genome shared. Alleles shared by relatives are identical by descent (IBD).
The coefficient of relatedness is expressed between 0 (non-relatives) and 1 (clones). Therefore, full siblings have a coefficient of relatedness of 0.5. See the wikipedia article on coefficent of relatedness for a table showing different types of relatives and their coefficient of relatedness.
The coefficient of relatedness (r) is expressed as the probability that two individuals will share the same allele due to recent common ancestry (e.g. they shared parents). It can also be thought of as the proportion of the genome shared. Alleles shared by relatives are identical by descent (IBD).
The coefficient of relatedness is expressed between 0 (non-relatives) and 1 (clones). Therefore, full siblings have a coefficient of relatedness of 0.5. See the wikipedia article on coefficent of relatedness for a table showing different types of relatives and their coefficient of relatedness.
Estimating relatedness coefficients
The relatedness coefficient of two individuals can be calculated two ways. One way to calculate it is if the pedigrees of the individuals are known, and is based on the number of steps in the pedigree between the two individuals. For more information on calculating relatedness by pedigree click here.
A lot of the time the pedigrees connecting two individuals are unknown. Genetic markers, such as the microsatellite data collected in Feathers for Science, can be used to estimate the relatedness between two individuals. For a given microsatellite "gene" an individual will have two different alleles, but across the whole population there may be many other possible alleles (e.g 35 different alleles) that occur at different frequencies.
The frequency of these alleles can be estimated from a population sample of genetic data. The frequencies of these alleles are informative to whether individuals are related (e.g. two individuals that share a very rare allele are more likely to be related).
A number of different mathematical methods have been developed to estimate relatedness using allele frequencies from multiple genetic markers. For individual x and y, these methods provide an estimate of relatedness (r) and 95% confidence interval - a range of values that the true r value could lie within. The range of possible values depends on variance in alleles that are identical by descent between two individuals (e.g. siblings may share more than 50% of their genome due to crossing over in meiosis), and possibility that alleles are identical by state. Identical by state means that the same mutation arose in the gene independently, and so are the same allele, individuals don't share the allele due to shared ancestry.
Participants have been provided with a relatedness estimate, and its 95% confidence interval. How was the best method chosen?
Each method has different strengths and weaknesses, but ultimately more genetic markers with more loci will provide more precise measures. To assess whether a method is right for the dataset at hand, it is recommended that the genetic data be used to simulate known relatives (e.g. parents and offspring, and full-siblings), and then use different methods to estimate the relatedness for each simulated pair.
Peri conducted the simulation of known relatives derived from the genetic data from the collected feathers. For each relationship type (parent-offspring, full-sibs, half-sibs and non-relatives), 100 pairs were simulated. She compared four different estimators (usually named for the inventor of the method), shown in Figure 3. The DyadML method is a more complicated maximum likelihood method that allows the calculation of relatedness values with and without inbreeding.The DyadML method, without accounting for inbreeding gave the best ability to discriminate between relatives, and between relatives and non-relatives.
A lot of the time the pedigrees connecting two individuals are unknown. Genetic markers, such as the microsatellite data collected in Feathers for Science, can be used to estimate the relatedness between two individuals. For a given microsatellite "gene" an individual will have two different alleles, but across the whole population there may be many other possible alleles (e.g 35 different alleles) that occur at different frequencies.
The frequency of these alleles can be estimated from a population sample of genetic data. The frequencies of these alleles are informative to whether individuals are related (e.g. two individuals that share a very rare allele are more likely to be related).
A number of different mathematical methods have been developed to estimate relatedness using allele frequencies from multiple genetic markers. For individual x and y, these methods provide an estimate of relatedness (r) and 95% confidence interval - a range of values that the true r value could lie within. The range of possible values depends on variance in alleles that are identical by descent between two individuals (e.g. siblings may share more than 50% of their genome due to crossing over in meiosis), and possibility that alleles are identical by state. Identical by state means that the same mutation arose in the gene independently, and so are the same allele, individuals don't share the allele due to shared ancestry.
Participants have been provided with a relatedness estimate, and its 95% confidence interval. How was the best method chosen?
Each method has different strengths and weaknesses, but ultimately more genetic markers with more loci will provide more precise measures. To assess whether a method is right for the dataset at hand, it is recommended that the genetic data be used to simulate known relatives (e.g. parents and offspring, and full-siblings), and then use different methods to estimate the relatedness for each simulated pair.
Peri conducted the simulation of known relatives derived from the genetic data from the collected feathers. For each relationship type (parent-offspring, full-sibs, half-sibs and non-relatives), 100 pairs were simulated. She compared four different estimators (usually named for the inventor of the method), shown in Figure 3. The DyadML method is a more complicated maximum likelihood method that allows the calculation of relatedness values with and without inbreeding.The DyadML method, without accounting for inbreeding gave the best ability to discriminate between relatives, and between relatives and non-relatives.

Figure 3: Box plot of relatedness values from simulation of known relative pairs using the allele frequency data derived from domesticated Gouldian finch feather samples. The title of each graph is the name of the relatedness estimator, including the DyadML estimator with and without accounting for inbreeding. The x-axis shows the simulated known relative pair (e.g. full-siblings), and the y axis shows the r-value estimated for each individual pair.
Some participants in Feathers for Science provided known relationships between individuals (e.g. individual with orange band is the mother of individual with the blue band). This allowed Peri to compare the performance of the chosen estimator (dyadML), and the data against known relatives (Figure 4). The estimates for full-siblings and half-siblings were combined because some participants did not provide the nature of sibling relationships, and it is not possible to know in an aviary with multiple breeding pairs.

Figure 4: Boxplot showing the estimated relatedness for simulated relatives, with known relatives provided by breeders in red. The simulated full and half siblings have been combined into a single category because of the difficulty of distinguishing full and half-sibs in a mixed aviary.
Based on Figure 4, these relatedness estimates for known parent-offspring pairs don't match well with their expectations (an r value of 0.5). There are several reasons why this might be the case a) Peri made a mistake in the lab, and the samples compared are not really relatives, b) The participants made a mistake and the original feather samples were incorrectly specified as relatives, or c) the amount of genetic data collected is not enough to be very accurate. The most likely situation is c.
In order to distinguish different types of relatives, typically 30-40 microsatellite markers are required (Blouin 2003). However, that number was inaccessible to Feathers for Science due to budgetary constraints. The relatedness estimates calculated here, however, can be useful for comparing whether certain groups (e.g. from breeder Joe Bloggs) are more related than expected by chance, and therefore the individual estimates do not need to be very accurate or precise.
Given these constraints, as a recipient of these data, you should interpret the r-values provided with a certain degree of skepticism. However, they may provide useful guidelines to inform you about which individuals are related, and which ones are not.
Inbreeding
Inbreeding describes when individuals with genotypes that are identical by descent (e.g. share a parent) mate and produce offspring. Because related individuals share many of the same genotypes, offspring will tend to be more homozygous (Figure 5). This can be a problem if there are deleterious recessive alleles in the population, which are more likely to be homozygous in the offspring of related individuals (Figure 5). This loss of fitness is referred to as 'inbreeding depression' and may cause death, disability, or loss of fertility.

Figure 5: Example of inbreeding and outbreeding on the genotypes of offspring. When the recessive alleles are deleterious, this can cause inbreeding depression.
Source: https://en.wikipedia.org/wiki/Inbreeding_depression
Inbreeding coefficients
Feathers for Science has provided two measures of inbreeding: individual inbreeding and population level inbreeding. These are explained below.
The individual inbreeding coefficient (f) refers to the probability that two alleles at a given locus (gene) were inherited from a common ancestor (i.e. alleles are identical by descent) (Wright 1921). Like relatedness estimators, these are measured out of 1. For example, the offspring from a parent-offspring mating has an inbreeding coefficient of at least 0.25. Similarly, the offspring from a half-sibling mating has a inbreeding coefficient of at least 0.125. If the parents were already inbred, the inbreeding coefficient for the offspring would be larger than if the parents weren't already inbred.
Like the relatedness estimators above, there are many mathematical ways to estimate this from genetic marker data. Essentially, they take population allele frequency information and the frequency of genotypes (i.e. heterozygotes and homozygotes) to estimate the inbreeding coefficient for each individual in a population.
The population level inbreeding coefficient (Fis) represents the average inbreeding within a population sample. This is measured by the ratio of observed heterozygotes vs the frequency of heterozygotes expected given the allele frequencies. The population level inbreeding coefficient (Fis) should be roughly similar to the average of the individual inbreeding coefficients (f).
The individual inbreeding coefficient (f) refers to the probability that two alleles at a given locus (gene) were inherited from a common ancestor (i.e. alleles are identical by descent) (Wright 1921). Like relatedness estimators, these are measured out of 1. For example, the offspring from a parent-offspring mating has an inbreeding coefficient of at least 0.25. Similarly, the offspring from a half-sibling mating has a inbreeding coefficient of at least 0.125. If the parents were already inbred, the inbreeding coefficient for the offspring would be larger than if the parents weren't already inbred.
Like the relatedness estimators above, there are many mathematical ways to estimate this from genetic marker data. Essentially, they take population allele frequency information and the frequency of genotypes (i.e. heterozygotes and homozygotes) to estimate the inbreeding coefficient for each individual in a population.
The population level inbreeding coefficient (Fis) represents the average inbreeding within a population sample. This is measured by the ratio of observed heterozygotes vs the frequency of heterozygotes expected given the allele frequencies. The population level inbreeding coefficient (Fis) should be roughly similar to the average of the individual inbreeding coefficients (f).
References
Blouin, M. 2003. DNA-based methods for pedigree reconstruction and kinship analysis in natural populations. Trends in Ecology & Evolution, 18: 503-511
http://dx.doi.org/10.1016/S0169-5347(03)00225-8
Miko, I. 2008. Mitosis, meiosis and inheritance. Nature Education, 1: 206
available at:
https://www.nature.com/scitable/topicpage/mitosis-meiosis-and-inheritance-476
Caldwell, J. 2014. Understanding genetics - relatedness.
available at:
http://genetics.thetech.org/ask-a-geneticist/inherited-parental-dna-two-half-siblings
Wright, S. 1922. Coefficients of Inbreeding and Relationship. The American Naturalist, 56: 330-338
https://doi.org/10.1086/279872
Wright, S. 1921. Systems of mating. Genetics 6:111–178.
Unknown Author. Unknown date. Calculation of the Coefficient of Relationship.
available at:
http://www.genetic-genealogy.co.uk/Toc115570135.html
